Molecular Chiroptical Properties
Chiral molecules are characterized by a unique three-dimensional handedness, and the resulting pairs of left- and right-handed enantiomers often exhibit distinct chemical activities when reacting within a chiral environment. Enantiomeric pairs of chiral molecules also exhibit distinct responses to left- and right-circularly polarized light in absorption, refraction, and scattering. These responses may be used to determine the handedness (i.e., the “absolute configuration”) of an enantiomerically pure sample, provided sufficient details about the corresponding circular dichroism, birefringence, or scattering intensity differences are known a priori.
|
The structure (left) and optical rotatory dispersion spectrum (right) of the helical molecule, (P)-[4]-triangulane.
|
![[4]triangulane structure](http://www.crawford.chem.vt.edu/wp-content/uploads/2019/03/triangulane_anim_small.gif)
![[4]triangulane ORD](http://www.crawford.chem.vt.edu/wp-content/uploads/2019/03/triangulane_ord.gif)
One of the major goals of our research group is the development of high-level ab initio quantum chemical models for prediction of molecular chiroptical properties such as specific rotation angles and CD spectra. Over the past several years, we have developed an efficient implementation of the coupled-cluster singles and double (CCSD) linear-response model that is applicable to medium-sized molecules such as [4]triangulane. These models are available as part of the PSI4 suite of quantum chemical programs. New developments attempting to extend the reach of these models to solvated systems (which are considerably more complex) has been a prominent feature in our current work.
Local Correlation Methods
The coupled cluster method is widely regarded as the “gold standard” of quantum chemical models because of the high accuracy it often provides for a variety of molecular properties, including structures, thermodynamic constants, and vibrational spectra. However, conventional coupled cluster theory suffers from high-degree polynomial scaling with the size of the molecular system (as measured by the number of electrons and the size of the one-electron basis set). The CCSD model, for example, scales as the sixth power of the molecular size, which implies that doubling the size of the molecule increases the computational time by a factor of 26 = 64. Even with state-of-the-art computing facilities, this scaling precludes application of coupled cluster theory to non-symmetric molecules larger than 10-12 heavy atoms.
Local correlation, a concept pioneered by Pulay and Saebø, provides one possible route over the scaling wall through a judicious choice of molecular orbital basis. The “canonical” MO’s, although convenient, are often delocalized over the entire molecular framework and often lead to overestimation of electronic interactions on spatially distant atoms. Pulay and Saebø demonstrated that if one abandons canonical orbitals and instead chooses a more localized form, vast numbers of electronic wave-function parameters become negligible and may thus be ignored.
|
Left: Contour representation of the delocalized highest-occupied molecular orbital of (R)-3-(1-Methyl-2-pyrrolidinyl) pyridine, also known as nicotine. Right: Distribution of the wave function amplitudes of dodecane by order of magnitude for both unperturbed and electric-field-perturbed wave functions. The sparsity of the perturbed wave function is reduced relative to the unperturbed state by more than an order of magnitude.
|
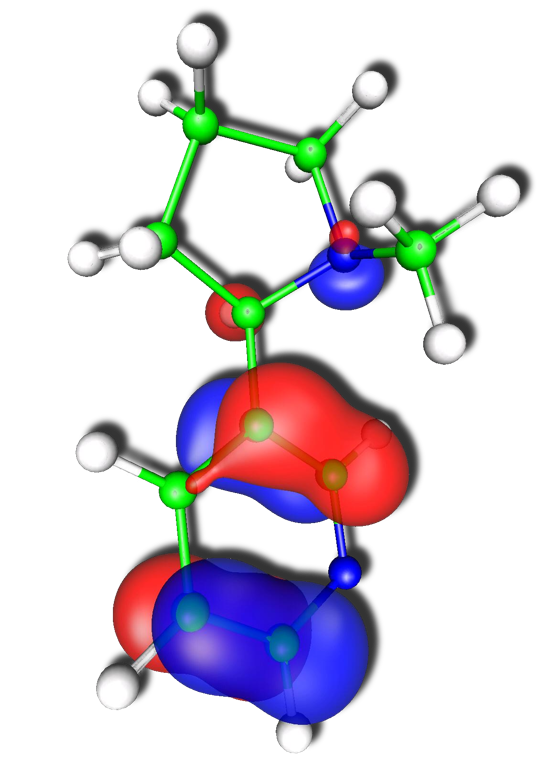
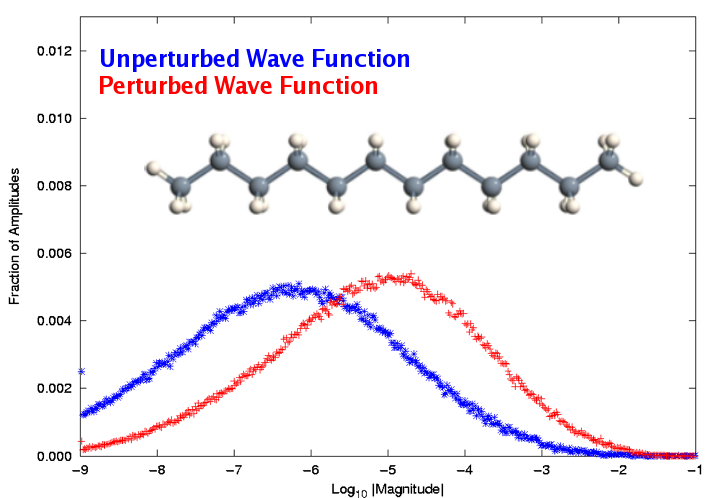
However, the application of the localization concept to molecular response properties introduces an additional complication: accurate representation of the derivative of the wave function with respect to an external perturbation, such as an electric or magnetic field, requires a more accurate representation of the unperturbed wave function than is necessary for computing the energy alone. Thus, the usual “orbital domain” structure that has performed so admirably for locally correlated ground-state energy calculations fails when applied to response properties. To compensate, we have devised a new domain construction approach based on analysis of the response of the individual molecular orbitals to the relevant external fields. This approach has proven successful for simple dipole polarizabilities, and work is underway to extend it to mixed electric/magnetic properties, such as optical rotation.
Sampling Configuration Space in Chiral Systems
Whether measured in solution or gas phase, the effects of conformational degrees of freedom are an important factor in the accurate prediction of chiroptical properties. In the gas phase, this can be accomplished by sampling a Boltzmann distribution of the main contributing conformers. A python-based driver for automating the process of conformation generation, Boltzmann population analysis, and ab initio property prediction using distributed computing resources is currently being developed. Results from this driver have been published in the Journal of Natural Products!
|
The structure (left), experimental (red) and theoretical electronic circular dichroism (middle) and UV (right) spectra of a chiral antimalarial trichospirolide.
|
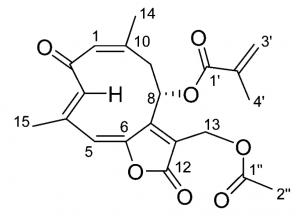
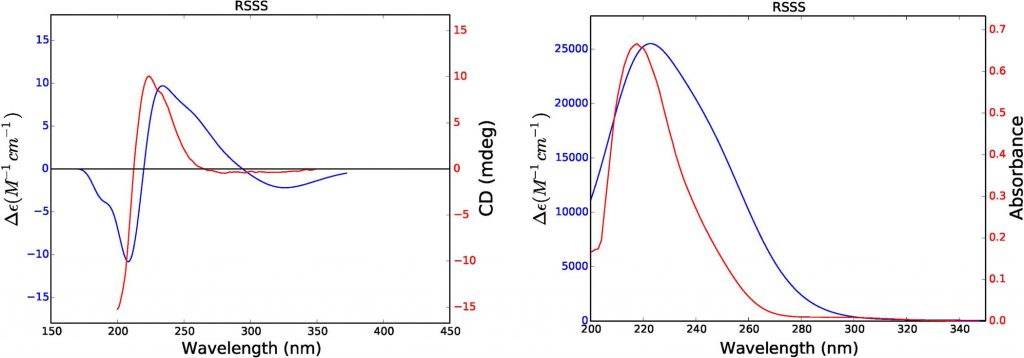
In solution phase, the configuration space is considerably larger due to the inclusion of solvent. It has been shown that small perturbations of the solvent shell can significantly impact chiroptical response; because of this, it is necessary to average over a sufficiently large sample of this space to accurately model the solute-solvent interaction. To this end, snapshots from molecular dynamics trajectories have been used to calculate the specific rotation of chiral solutes in different solvents. A paper on this work is currently being revised.
Alternative Models in Electronic Structure Theory
In addition to pushing the boundaries of traditional electronic structure methods like coupled cluster and density functional theory, additional models are being considered. These include Monte Carlo simulations, fragmentation schemes, real-time methods (such as real-time Coupled Cluster), and machine learning. Check back for more details on these and other projects as they develop.